






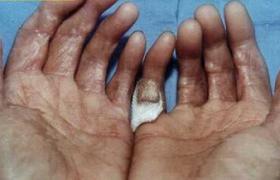
擠奶人結節
来源:74U閱讀網

糖原累積癥
糖原累積癥百科
糖原貯積癥是少見的一組相關的常染色體隱性遺傳病,患者不能正常代謝糖原.因此糖原(一種淀粉)大量累積.最嚴重的糖原貯積癥是糖原貯積癥Ⅱ型(龐珀病),通常在1歲內發病.糖原蓄積於肝、肌肉、神經及心臟,使其不能正常工作.舌、心、肝長大.患兒如嬰兒般軟弱且進行性無力,有呼吸及吞咽困難.龐珀病是不可治愈的,大多數患兒2歲時死亡.不嚴重的龐珀病可累見於年長兒童及成人,造成上、下肢肌無力及深呼吸功能減退.其他類型糖原貯積癥患者有痛性痙攣及肌無力,通常出現於運動後,癥狀輕重不一.避免運動可使癥狀消退.肌肉的損害導致肌球蛋白釋放入血.由於肌球蛋白從尿排泄,因此可用尿檢查,診斷糖原貯積癥.肌球蛋白對腎臟有害,限制運動可降低肌球蛋白水平,大量飲水,尤其在運動後,可稀釋肌球蛋白.肌球蛋白水平高時,可用利尿劑防止腎臟損害.肝移植對糖原貯積癥有效但對龐珀病無效.

糖原累積癥
糖原累積癥病因
GSD是由於患者缺乏糖原代謝有關的酶,使糖原合成或分解發生障礙,導致糖原沉積於組織中而致病,由於酶缺陷的種類不同,造成多種類型的糖原代謝病,其中Ⅰ,Ⅲ,Ⅵ,Ⅸ型以肝臟病變為主,Ⅱ,Ⅴ,Ⅶ型以肌肉組織受損為主.
糖原貯積病系遺傳性糖原代謝紊亂,其發生率為1/2萬 依其酶缺陷(多數屬分解代謝上的缺陷)的不同可分為12型 除磷酸化酶激酶缺乏外, 均為常染色體隱性遺傳病.患者多於嬰幼兒發病死亡.
病因:
糖原貯積病為常染色體隱性遺傳,磷酸化酶激酶缺乏型則是X-性連鎖遺傳 .
發病機制:
糖原貯積病系遺傳性糖原代謝紊亂.
糖原在機體的合成與分解是在一系列的酶的催化下進行的,當這些酶缺乏時, 糖原難以正常分解與合成,累及肝、腎、心、肌肉甚至全身各器官,出現肝大、低血糖、 肌無力、心力衰竭等.

糖原累積癥
糖原累積癥癥狀
糖原貯積病主要表現為肝大、低血糖,包括Ⅰa型(葡萄糖-6-磷酸酶缺乏)及更罕見的Ⅰb型(G-6-P微粒體轉移酶缺乏)、Ⅲ型、Ⅵ型和伴X染色體與常染色體隱性遺傳的磷酸酶b激酶缺乏.肌-能量障礙性糖原貯積病主要表現為肌肉萎縮、肌張力低下、運動障礙,包括Ⅴ型、Ⅶ型,磷酸甘油變位酶缺乏和LDHM亞單位缺乏另有Ⅱ型、Ⅳ型等.

糖原累積癥
糖原累積癥檢查
實驗室檢查:
1.空腹血糖測定.
2.血總膽固醇、叁酰甘油測定.
3.血乳酸測定、尿酸測定.
4.胰高糖素試驗.
5.肝功能轉氨酶測定.
其他輔助檢查:依據病情應選做骨骼X線檢查、腹部B超、心電圖、超聲心動圖等.必要時做組織或器官病理活檢.
糖原累積癥預防
糖原貯積病系遺傳性糖原代謝紊亂,無明確相關預防資料.其發生率為1/2萬.依其酶缺陷(多數屬分解代謝上的缺陷)的不同目前可分為12型.除磷酸化酶激酶缺乏外,均為常染色體隱性遺傳病.患者多於嬰幼兒發病死亡.
治療主要是延緩病情的發展,增加肌力,改善癥狀及改善呼吸苦難,延長生命,改善生存質量,減輕痛苦.