





面肌痙攣
来源:74U閱讀網

法佈裡病
法佈裡病百科
法佈裡病(Fabrydisease)或安德森-法佈裡病(Andeson-Fabrydisease),或α-半乳糖苷酶A缺乏病(alpha-galactosidaseAdeficiency),於1898年首先由英格蘭的安德森和德國的法佈裡幾乎同時報道.法佈裡將該疾病的皮膚表現稱為彌漫性體血管角質瘤.

法佈裡病
法佈裡病病因
一、發病原因
本病為性連鎖顯性遺傳疾病,致病基因定位於X染色體長臂中段Xq21至Xq24.因此,半合子男性病情重,而雜合子女性病情表現多樣,多數輕度異常(如僅有角膜環生狀混濁),甚至無癥狀,少數重如男性.該基因突變導致α-半乳糖苷酶A(一種溶酶體水解酶)缺乏,使終端α-半乳糖殘基不能從中性糖鞘脂(neutral glycosphingolipid)分子上解離,而使如此的中性糖鞘脂,主要為酰基鞘氨醇三己糖苷(ceramide trihexoside),也有較少的酰基鞘氨醇三己糖苷沉積至體內各器官致病.臨床發現B型或AB血型的Fabry病患者常發病早、病情重,這是因為這兩種血型人群還具有另外兩個糖鞘脂--B和B1糖鞘脂,當α-半乳糖苷酶A缺乏時,它們亦不能解離掉分子終端的α-半乳糖殘基,該B及B1糖鞘脂也一同沉積至器官組織.由於B或AB血型患者比O或A血型患者有更多的糖鞘脂沉積,故病情重.
二、發病機制
1967年Brady等闡明瞭該病的生物化學缺陷.他們認為溶酶體酶酰基鞘氨醇己三糖苷酶活性缺陷導致瞭神經氨醇分解代謝減低,從而導致細胞內神經氨醇半乳糖苷酰基鞘氨醇的沉積.上述細胞內沉積物a末端連接一個半乳糖苷殘基.由於人們對這一特異酶缺陷的認識,現在可以準確地診斷半合子的男性,並可以辨認雜合子女性攜帶者以及在宮內受到影響的胎兒.
現已明確,血管角質瘤綜合征是一種X連鎖遺傳的疾病,缺陷基因位於X染色體長臂上.半合子中該基因的外顯率很高,酶缺陷的臨床表現既有傢族內也有傢族間變異.α-半乳糖苷酶的整個基因組序列都已被破譯,而且可以得到足夠長度的cD-NA.不同傢族分子排列不同,存在著外顯子突變、基因重排和堿基對缺失.受影響的半合子男性表現出多系統臨床表現,雜合子的女性則有著不同的臨床表現,也可無癥狀,但可找到脂質貯積的證據.
(溫馨提示:以上資料僅提供參考,具體情況請向醫生詳細咨詢.)

法佈裡病
法佈裡病癥狀
一、癥狀
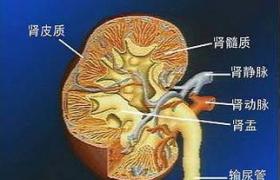
1、腎臟表現腎臟受累主要表現為高血壓、血尿、蛋白尿和脂肪尿,50%患者出現水腫.腎小管功能不全如濃縮稀釋、酸化功能等障礙是本病的早期表現.隨病情進展,30%患者在20~40歲最後進入終末期出現腎衰竭,此時腎臟體積變小.如腎血管病變比腎小球病變嚴重可並發腎梗死.腎臟病變是法佈裡病發病和死亡的首要原因.心血管病和腦血管病也是本病常見表現.腎臟病變最常表現為20~30歲間出現的隱匿輕微蛋白尿(0.5~2.0g/24h).30~50歲常發展至尿毒癥伴高血壓,也有患者早在10~20歲發展至終末期腎臟病.一般腎病范圍的蛋白尿不常見.無蛋白尿的患者在偏振光顯微鏡下可見尿中馬耳他十字或在亮區顯微鏡下發現卵圓形的脂質體,提示存在脂質貯積病的可能.如在尿中發現髓磷脂體可確診法佈裡病.雜合子(女性)和半合子(男性)患者尿中神經鞘脂的含量比正常人多20~80倍.患者可有輕微的鏡下血尿.由於該疾病以X連鎖的隱性方式傳遞,半合子腎功能衰竭很常見.而雜合子也可發展至終末期腎臟病.
二、系統的表現
(1)皮膚血管角質瘤:是本病特征性損害,發病率約90%,平均發病年齡17歲.以陰囊部角化血管瘤最為明顯,常伴有面部毛細血管擴張,在皮膚超表層呈簇或葡萄狀的點狀的鮮紅、紫紅或紅黑色的靜脈血管擴張區,有出血傾向,壓之不褪色,較大皮疹可有過度角化.系皮膚血管的內皮細胞與平滑肌因鞘糖脂的沉積而弱化後擴張而來.出生時僅可見,以後可擴展成4mm,可高出表面,分佈於臍膝之間的所謂“坐浴區”(軀幹下部、臂、股、髖部及會陰處),常兩側對稱.隨年齡增長,角質瘤的數量與面積也增多增大.
(2)自主神經系統功能失常:神經系統表現常是本病最早出現的癥狀,發病年齡在10歲,最初表現可發生在年僅5歲的小兒,可先於血管角質瘤多年前出現,故兒科醫師對此現象的認識十分必要.
血管角質瘤綜合征神經系統受損的表現,主要是發作性痙攣掌痛(Fabrys危象)與四肢蟻爬感.Fabrys病患者痙攣掌痛與四肢蟻爬感典型表現是在冷熱、運動、勞動後,手掌和足底間歇發作性刺痛、燒灼痛,向四肢近端放射,嚴重的周期性發作持續數分鐘到數周.呈周期性發作疼痛者為77%,慢性病程者為89%,終身持續者為90%.也可表現為雷諾征、腹痛.隨年齡增長疼痛發作次數減少、程度減輕.誘發因素有發熱、天氣變暖、運動、緊張、飲酒.疼痛程度劇烈時,常伴有疲乏無力、發熱、出汗與血沉增快而誤為風濕.查體無神經系統體征.
此外,尚有37%患者有中樞神經系統損害癥狀,發病年齡一般大於26歲,表現為腦卒中(占24%),癡呆、被動和壓抑社會交往活動障礙等人格改變(占18%).腦脊液檢查正常,腦部磁共振成像(MRI)檢查可早期發現白質和灰質小的病灶.
自主神經功能受損可以出現少汗或無汗、縮瞳、淚液與涎液減少、陽萎與直立性低血壓等,出現胃腸道癥狀者約有69%,表現為餐後發作性腹痛、腹瀉、惡心嘔吐,脂肪不能耐受等,多數患者明顯消瘦.
(3)眼部病變:眼部體征是Fabrys病具有特征性變化之一.角膜混濁可見於所有的雜合子和大多數的半合子.患者可表現有晶狀體前部和後部的異常,出現白內障及視網膜血管迂曲、擴張,角膜混濁、角膜漩渦狀沉積物,盡管這不影響視力或視力輕度減退,特別是女性患者70%~80%可以僅表現出該病的眼部體征,當眼科醫師發現患者角膜變性或者晶狀體的改變時,應該想到Fabrys病,並建議患者去做基因檢查,以便發現該患者的其他傢庭成員是否也患有此病.因此,眼科醫師在Fabrys病的診斷中應起著非常重要的作用.
(4)心臟損害:心臟受損常常是Fabrys病患者的死因之一.主要表現為傳導障礙,心肌病,冠狀動脈功能不全或冠狀動脈阻塞導致心肌梗死,高血壓(腎臟缺血導致腎素分泌增加),瓣膜與升主動脈退行性病變(二尖瓣脫垂多見).一般來講,Fabrys病患者溶酶體酶α-半乳糖苷酶A酶活性僅有正常人的1%~17%,如果殘餘酶活性高,患者可以無癥狀或僅出現心臟的病變.當引起缺血性心臟病時患者可出現心絞痛、心肌梗死和充血性心力衰竭而死亡.
(5)其他系統器官:血管角質瘤綜合征患者除以上臨床表現外,還可以見到進行性感覺神經性聽力喪失(78%);面部出現水腫,嘴唇增厚,唇皺褶增多畸形(56%);此外,部分患者可出現溶血性貧血、淋巴結病、肝脾腫大、骨無菌性壞死、肌病、肺功能減退、免疫功能低下,血小板聚集增強而易發生血栓與栓塞.
本病雖有許多特征性表現,但臨床誤診10年以上者並不少見.一般來講,Fabrys病患者的發病年齡在兒童晚期或青少年早期,男性發病,女性為攜帶者或輕癥患者,B型和AB型血型者發病早且較嚴重.患者平均存活年齡為50歲,女性基因攜帶者的生存年齡較長,約70歲.死因主要是腎衰或心腦血管並發癥.
由於法佈裡病患者神經酰胺三己糖苷的正常分解代謝所需的溶酶體酶α-半乳糖苷酶A功能缺乏或活力降低,糖鞘脂代謝存在障礙,致使脂質分解代謝過程中神經鞘脂類,主要為腦胺三已糖苷(ceramidetrihexoside,CTH)在全身各種組織中廣泛沉積,如血管內皮與平滑肌的細胞內、中樞神經系統神經元、周圍神經系統神經節、皮膚、眼、胃腸道、心臟、腎臟等,因此臨床癥狀表現為多系統損害,有時以某一系統癥狀為主.通常依據臨床表現、特征性體征以及陽性傢族史,活檢組織以及培養的皮膚成纖維細胞內α-半乳糖苷酶活性低下等明確診斷並不困難.如果臨床表現提示為α-半乳糖苷酶A缺陷,通過測定從外周血分離出來的白細胞中該酶的濃度可助診斷.半合子患者幾乎無酶的活性.如果該酶活性達正常人6%~20%,則可無臨床癥狀.在雜合子,該酶活性水平介於正常人和半合子之間.測定白細胞中α-半乳糖苷酶A活性不是辨別攜帶者的敏感方法,辨別攜帶者最好是測定尿中酰基鞘氨醇雙半乳糖化物和三羥化物濃度.測定培養的羊膜細胞α-半乳糖苷酶A水平可進行產前診斷.
隻有很少的貯積疾病有與法佈裡病一樣的腎臟病變和貯積物的分佈.腎小球小管上皮細胞空泡形成無特異性,但包涵體超微結構特點有診斷意義.在考慮為本病且有癥狀的男性患者,如包涵體分佈廣泛且受累細胞中包涵體數目眾多,尤其是在腎小球的上皮細胞中,應考慮本病診斷.尿沉渣發現含有典型包涵體和遊離髓磷脂體的完整細胞,對診斷有重要價值.
(溫馨提示:以上資料僅提供參考,具體情況請向醫生詳細咨詢.)

法佈裡病
法佈裡病檢查
一、檢查
1、尿液檢查常規檢查可見血尿、蛋白尿和脂肪尿,腎小管功能不全如濃縮稀釋、酸化功能障礙等.尿沉渣檢查可見有脂質包涵體的細胞,超微結構檢查發現含有典型包涵體和遊離髓磷脂體的完整細胞.仔細鏡檢尿沉渣,常可見內含雙折光脂質的泡沫樣上皮細胞(此脂質細胞在偏光鏡下形似馬耳他十字架),對本病具有提示意義.
2、血液生化檢查血漿、白細胞或成纖維細胞中的α-半乳糖苷酶活性明顯降低.其原理是由於溶酶體酶α-半乳糖苷酶A將酰基鞘氨醇水解為鞘氨醇和一個自由脂肪酸,當缺乏該酶時糖鞘脂在血漿中的濃度和組織中的沉積增多.半合子患者血漿中酰基鞘氨醇三羥化物濃度增高可達正常值3倍,在一些組織中增多可達300倍.
3、羊膜或絨毛囊穿刺通過檢測羊水及絨毛中(妊14周時)α-半乳糖苷酶的活性有助於產前診斷.
腎活檢病理檢查:
1、光鏡檢查光鏡下腎小球臟層上皮細胞(有時還有壁層上皮細胞、內皮細胞及腎小管上皮細胞)增大,含無數空泡,空泡中充滿脂質,偏光鏡下見雙折光脂質體,形似馬耳他十字架;腎小球壁局灶性增厚,腎小球周圍及間質纖維化.
本病腎小球變化最顯著,石蠟切片顯示腎小球臟層上皮細胞擴大,細胞內空泡形成,空泡通常小而均一,故細胞呈蜂窩狀,胞漿幾乎消失而不宜辨認,壁層上皮細胞受累相對少見,內皮細胞和系膜細胞偶可見到空泡.在早期或較輕的患者,腎小球可無其他異常,隨著病情進展至腎功能衰竭,逐漸進展至腎小球節段性或全球性硬化.在硬化小球仍能發現有空泡形成的細胞,為進展性腎病患者的診斷提供瞭線索.腎小管有相似空泡細胞,以遠曲小管和亨勒(Henle)襻最為明顯.近曲小管細胞較少受累.動脈及小動脈中有大量空泡細胞.內皮細胞常受累,細胞擴大呈泡沫狀.血管中層平滑肌細胞內有大小不等的空泡.
鋨固定塑料包埋組織切片用甲苯胺或亞甲藍處理後,可在先前提到的細胞內包涵體的相應部位見到細的暗染的顆粒狀包涵體.這種技術診斷價值更大,尤其是在完全或局灶硬化的腎小球,這種方法也可以用來辨認受累的間質細胞.
2、免疫熒光檢查免疫熒光檢查常為陰性.發展至晚期損害的腎小球,如節段性硬化時,可出現IgM、C3和C1q呈節段性或顆粒狀分佈於血管壁或系膜區.
3、電鏡檢查電鏡下顯示上述細胞包含無數圓或卵圓形層狀小體(這些小體被稱為“斑馬小體”或“髓鞘脂影”),對本病診斷很有意義.
本病幾乎所有腎臟細胞均可受累,以腎小球毛細血管上皮細胞最甚,受累細胞溶酶體內可見典型的同心圓排列的層板樣異常包涵物,為電子致密的嗜鋨性層狀小體,由於切面不同可呈太陽光環狀、蠕動的絨毛狀或斑馬紋狀結構.多種細胞胞質內有多種形態擴大的溶酶體,呈洋蔥皮樣、層板樣、致密實心球狀或空泡狀,因此該病的溶酶體改變亦可呈多樣性.此外,還可有上皮細胞足突融合,基膜灶性增厚等改變.
二、鑒別
在疾病的早期或癥狀不典型時,應與以下疾病相鑒別:
1、風濕病風濕熱臨床較為常見,一般有先期的鏈球菌感染史,抗“O”增高、皮下結節、關節炎及舞蹈病等癥狀體征,抗風濕治療有效.
2、藥物性眼損害藥物性眼損害有明確的服藥史,如氯喹可引起與Fabrys病相似的角膜混濁現象.
3、心腦血管性疾病年輕人出現嚴重的痛性神經病變或有抽風、偏癱、人格與行為改變,伴進行性腎、心血管和腦血管的功能障礙,應想到本病,MRI可早期發現腦損害.
此外,註意與其他原因引起的腎小球腎炎和腎小管功能障礙的疾病相鑒別.
(溫馨提示:以上資料僅提供參考,具體情況請向醫生詳細咨詢.)
法佈裡病預防
一、預防
本病目前無有效預防措施,積極對癥治療可有效控制病情進展,並預防早期發生腎衰竭.
二、護理
(溫馨提示:以上資料僅提供參考,具體情況請向醫生詳細咨詢.)